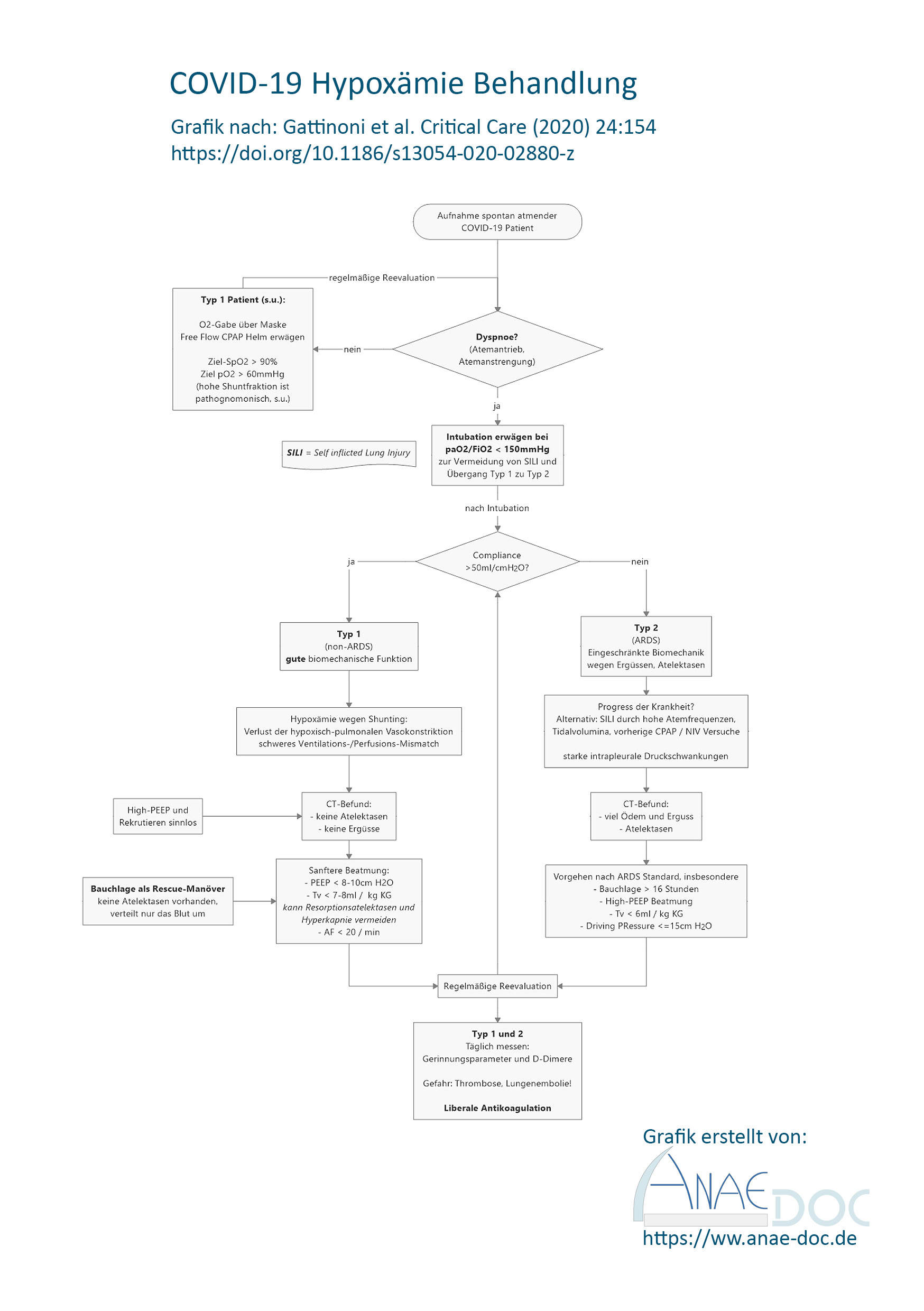
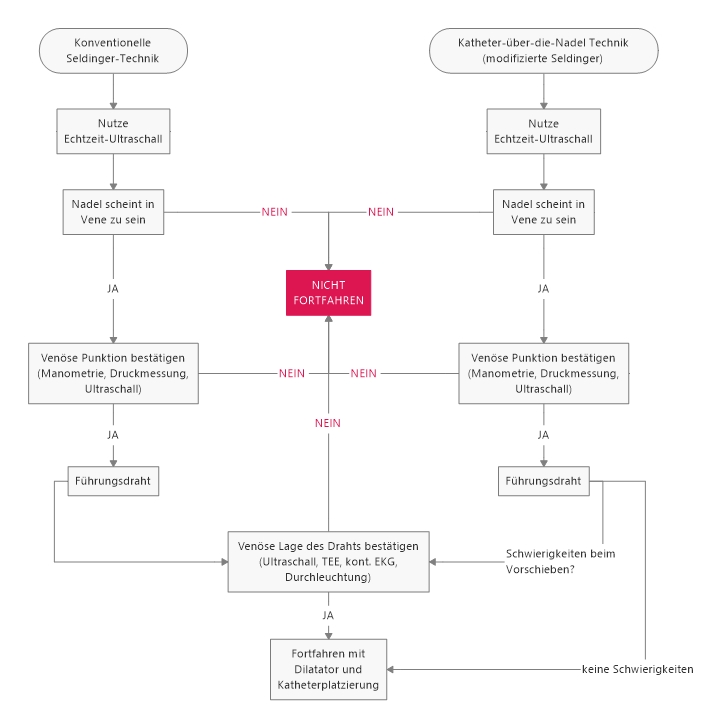
Heute möchte ich etwas zum Thema Sichelzellkrankheit schreiben. Wohlgemerkt „-krankheit“, weil es viel mehr ist, als bloß die Anämie. Zumindest in meinem Studium hätte der Eindruck entstehen können, dass es nicht mehr ist. Aber dazu im Folgenden mehr.
Der Krankheit liegt die Mutation des Gens für die beta-Kette des Hämoglobins zugrunde. Zur Erinnerung: HbA (adultes Hämoglobin) besteht aus zwei alpha- und zwei beta-Ketten. Das erklärt auch, warum die Erkrankung erst mit etwa 6 Monaten „beginnt“. Das fetale Hämoglobin besteht aus 2 alpha- und 2 gamma-Ketten. Erst wenn die beta-Ketten gebraucht werden, stellt sich heraus, dass da ein Fehler vorliegt.
Mischformen mit anderen Anämie-Arten sind möglich: Zum Beispiel die Kombination aus Sichelzellneigung und Thalassämie. Phänotypen können heißen: HbSb-Thalassämie, HbSD, HbSO, HbSE etc.
Im angloamerikanischen Raum ist ein Neugeborenen-Screening darauf etabliert; im deutschsprachigen Raum leider trotz aller Bemühungen immer noch nicht. Immerhin ist die Prävalenz hier sehr gering.